Poster
in
Workshop: Machine Learning in Structural Biology
SPRINT Enables Interpretable and Ultra-Fast Virtual Screening against Thousands of Proteomes
Andrew McNutt · Abhinav Adduri · Caleb Ellington · Monica Dayao · Eric Xing · Hosein Mohimani · David Koes
{kind=link}
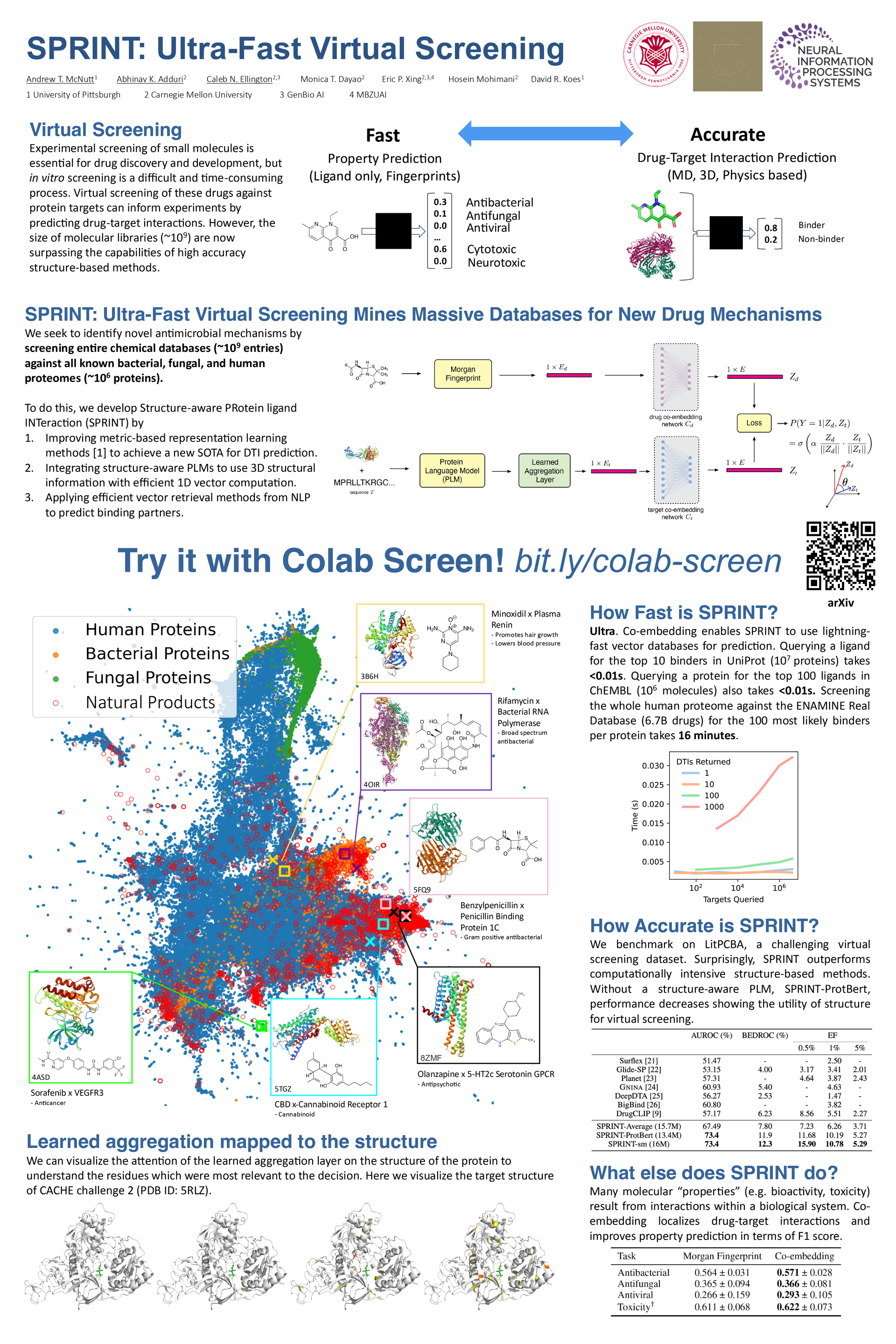
Virtual screening of small molecule drugs against protein targets can accelerate drug discovery and development by predicting drug-target interactions (DTIs). However, structure-based methods like molecular docking are too slow to allow for broad proteome-scale screens, limiting their application in screening for off-target effects or new molecular mechanisms. Recently, vector-based methods using protein language models (PLMs) have emerged as a complementary approach that bypasses explicit 3D structure modeling. We introduce SPRINT, a vector-based approach to screen entire chemical libraries against whole proteomes for DTIs and novel mechanisms of action. SPRINT improves on prior work by using multi-head attention pooling and structure-aware PLMs to learn protein embeddings, achieving SOTA in virtual screening on the LIT-PCBA dataset and DTI prediction benchmarks, while providing interpretability in the form of residue-level attention maps. SPRINT uses vector databases to achieve extremely fast runtimes: querying the whole human proteome against the ENAMINE Real Database (6.7B drugs) for the 100 most likely binders per protein takes 16 minutes. We anticipate that SPRINT will enable screening chemical libraries against the human proteome for efficient drug repurposing. We further plan to use SPRINT to simultaneously screen against thousands of microbial proteomes to identify promising antimicrobials with novel mechanisms and minimal off-target effects in humans.