Poster
in
Workshop: NeurIPS 2024 Workshop: Machine Learning and the Physical Sciences
Fine-tuning Foundation Models for Molecular Dynamics: A Data-Efficient Approach with Random Features
Pietro Novelli · Luigi Bonati · Pedro J. Buigues · Giacomo Meanti · Lorenzo Rosasco · Michele Parrinello · Massimiliano Pontil
{kind=link}
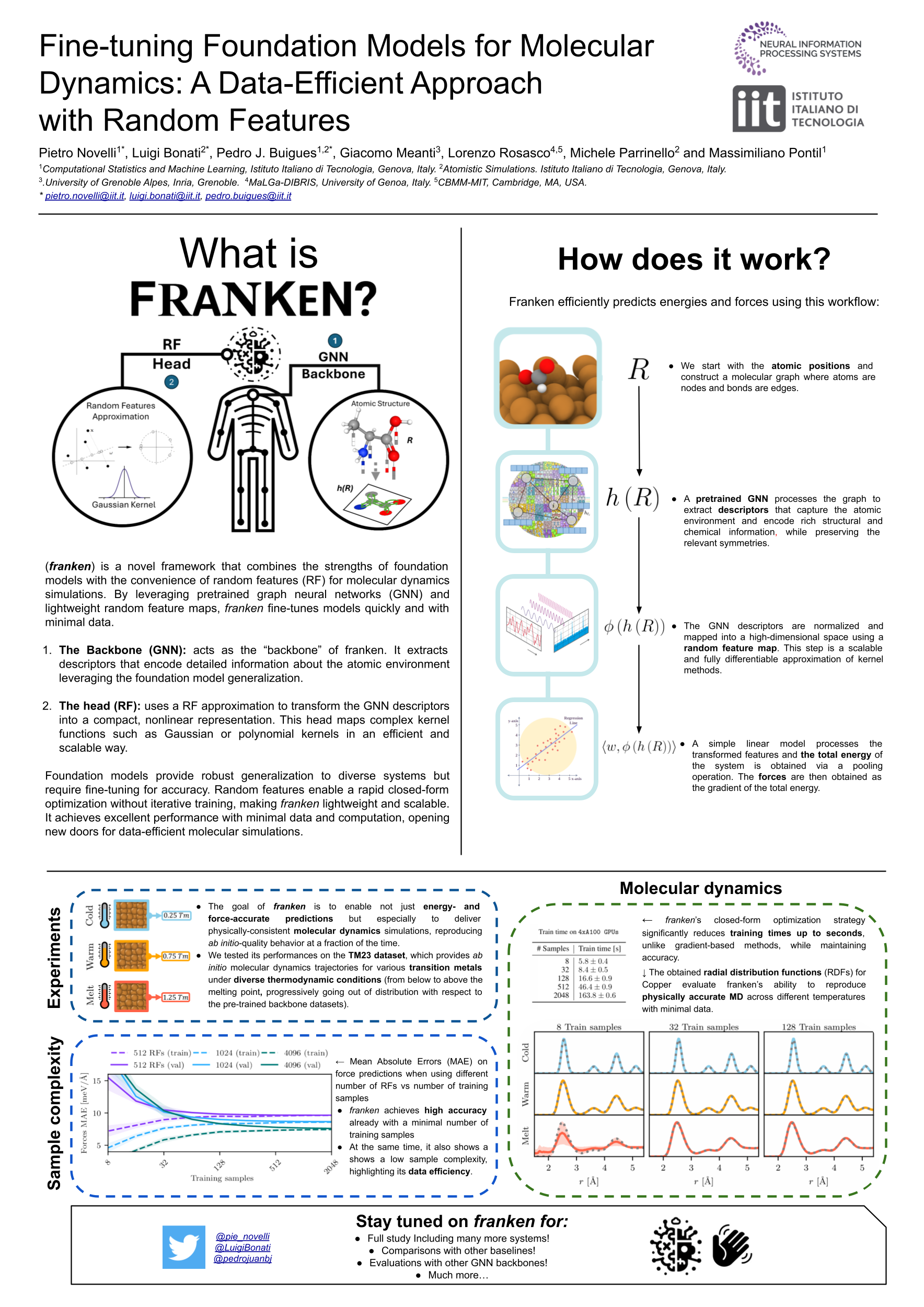
Accurate modeling of atomistic interactions using machine learning (ML) interatomic potentials has become an essential tool for molecular dynamics simulations. However, training these models typically requires large amounts of expensive ab initio data, such as those generated by density functional theory (DFT). Recently, foundation models trained on large and diverse datasets have shown promise because of their good performance, even on out-of-distribution systems. Despite this progress, they are still far from optimal and often require further fine-tuning. Doing so, especially in a data-efficient and computationally feasible way, remains a key challenge. In response, we present franken, which combines a representation extracted from graph neural networks (GNNs) with random features (RFs) models. Through experiments on systems from the TM23 transition metals dataset, we show that franken provides accurate and robust molecular dynamics simulations with minimal sample complexity, providing an efficient path to high-quality results.